WP2: Biophysical experimentation
- Alison Edwards (Deactivated)
WP2 Biophysical experimentation (PDRA2,3) develops and applies innovative biophysical technologies to the biological components of WP1 to determine the molecular architecture of functional pyrenoids, how function is linked to composition and physicochemistry, and how these properties depend on internal constituents and external microenvironment. WP2 generates high-
precision localisation and kinetics data that feeds into the modelling framework of WP3.
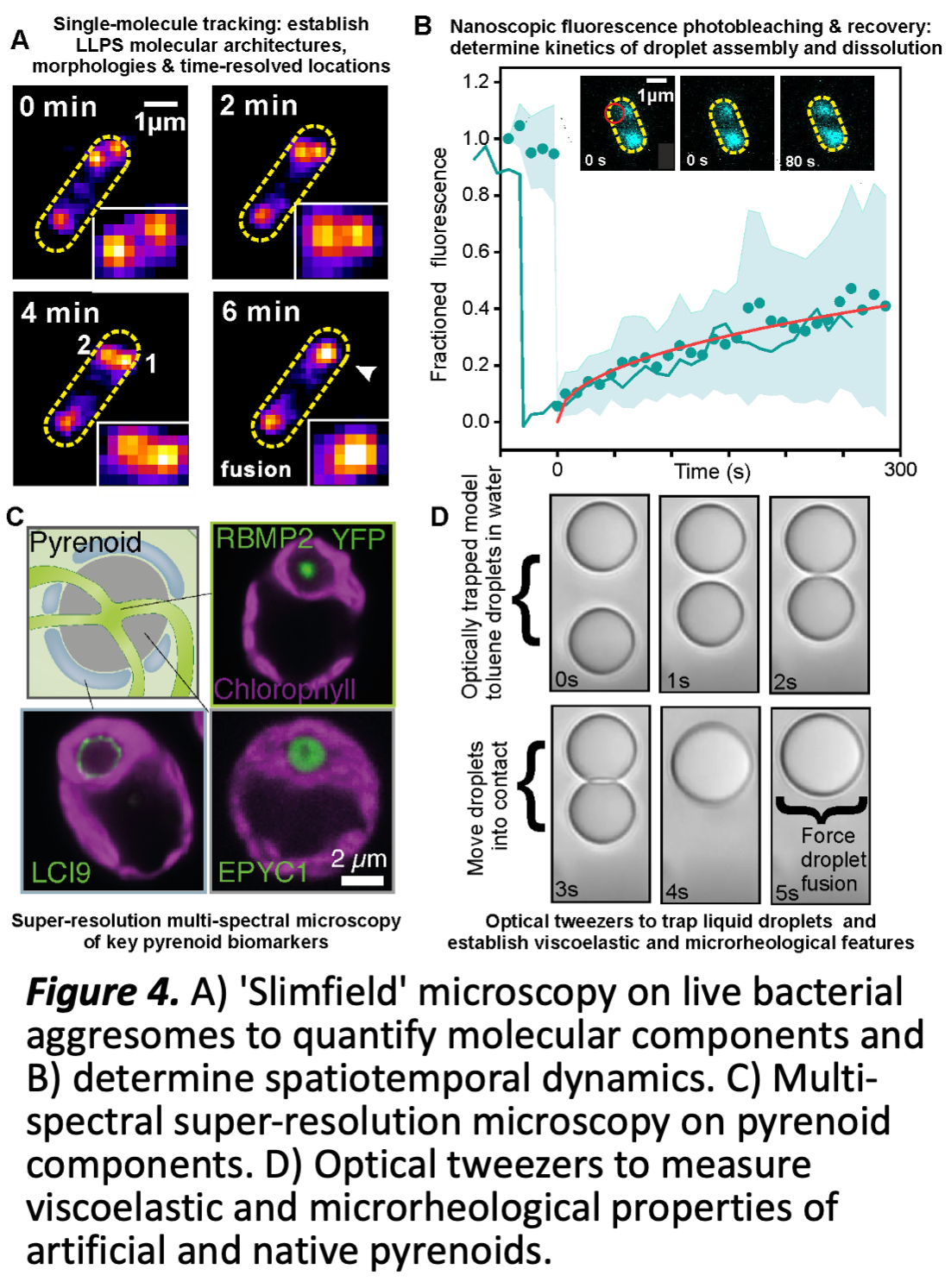
WP2 Preliminary work has made significant progress in purifying and fluorescently labelling several key pyrenoid components to develop reconstituted pyrenoid liquid droplets in vitro and establishing robust protocols for production of Chlamydomonas and Chlorella Rubisco and EPYC1 (Fig 3). We have also developed a robust experimental pipeline for characterising dynamic nanoscopic LLPS compartments in live cells, optimised via our research into bacterial aggresomes (Fig 4 A,B) [10]. Promising super-resolved preliminary data uses a range of key pyrenoid fluorescent reporters in live algae (Fig 4C), and bespoke optical trapping (Fig 4D).
WP2 Programme of work
WP2.1 In vitro quantification of pyrenoid protein interactions (MP; M1-M36) focuses on two systems (C. sorokiniana, C. reinhardtii), chosen for their differing pyrenoid structures. WP2.1 will map phase diagrams of Rubisco/linker proteins interactions in vitro in these two systems using microscopy and spectrophotometry. Initial experiments will determine how Rubisco/linker protein-ratios and solution pH (known to correlate with in vivo pyrenoid activity) affect LLPS, with salt and redox potential then explored. Results will drive and interrogate phase models (WP3), and set conditions for more intensive cellular (WP1), biophysical and structural analyses (here, WP2.2). Shape and dynamics of condensates will be measured in vitro by (fluorescence) microscopy. Binding kinetics and affinities for Rubisco/linker protein interactions will be measured using surface plasmon resonance or isothermal titration calorimetry. NMR spectroscopy will be used to characterise structure and dynamics of disordered linker proteins, as well as hydrodynamic properties (e.g.
diffusion constants) of components in different phases. The published structure of the C. reinhardtii Rubisco-EPYC1 complex [4] identifies interaction sites on Rubisco and will be used to interpret data collected on this system and C. sorokiniana. Driven by results from WP1 and 3, mutant linker proteins will be designed (e.g. modifying number, spacing or composition of RBMs) and their biophysical and phase separating properties characterised, and these data will be used to refine bioinformatic analysis in WP1 and system properties in WP3.
WP2.2 Complexing components (MP; M18-M36) evaluates protein LLPS in the presence of membranes and starch plates. WP2.2 examines linker proteins tethered to an artificial membrane system, testing micelles, liposomes or lipidic cubic phases, and monitors LLPS via fluorescence microscopy. Starch plates will be purified from native sources using established protocols, combined with starch-matrix protein tether(s) and added to in vitro LLPS assays. The complex LLPS constructs evaluate multiscale models of WP3.2 and WP3.3, informed by the data from WP2.3/2.4 (below).
WP2.3 interrogates the molecular composition of pyrenoids, their architecture and dynamics, and the influence of the microenvironment (ML; M1-M24) ML has developed an advanced super-resolved bioimaging pipeline (Fig. 4B) using rapid millisecond single-molecule Slimfield microscopy to monitor the spatial localization, stoichiometry and dynamics of multiple fluorescently labelled components of biomolecular condensates in live cells. WP2.3 applies this pipeline to in vitro reconstituted pyrenoids, existing algal cell lines and those newly developed from WP1. It will use multispectral imaging in these strains to determine the spatial distribution of different pyrenoid components using nanoscale localization microscopy tracking of fluorescently labelled components, establishing what components are interacting through precise colocalization analysis and how these are structured into pyrenoids. Precise 'stoichiometry' for each component will be determined using advanced image analysis software ML has developed to count molecules in tracked fluorescent foci [11]. Using in vitro formulations of increasing complexity as steered by the outcome of WP1, we will map the molecular architecture of the Rubisco-linker matrix, the membrane-matrix interactions, and the matrix-starch interactions. Rapid sptPALM with photoswitchable fluorescent dye labels, will establish the mobility of key components inside both synthetic and native live cell pyrenoids, adapting and improving previously developed Bayesian inference algorithms [12] to categorise different modes of
pure diffusion vs. reaction-diffusion to quantify the precise location of reaction events and their associated rate constants, and compare with simulations from WP3. This analysis will be complemented by fluorescent recovery after photobleaching (FRAP) experiments to establish the parameters for molecular flux and turnover processes detected between pyrenoid and the surrounding matrix. New multi-spectral FRAP capability will determine whether molecular components turnover cooperatively or independently to determine the physical rules of dynamic exchange inside pyrenoids. We will use a range of FRET based reporter probes optimised previously in budding yeast to quantify how these molecular architecture and turnover dynamics are correlated to the local physicochemical environment surrounding pyrenoids utilising microfluidics and temperature control capability to precisely control the sample, such as molecular crowding, viscosity, ionic strength and pH, providing rich multilevel data for WP3.
WP2.4 explores how the microrheological properties of pyrenoids constrain shape, spatial patterning and function (ML; M12-M36) The size and shape of pyrenoid droplets in live cells does not follow traditional expectations of classical nucleation for liquid-liquid phase separation: instead of growing continuously, a preferred length scale emerges, suggesting more complex underlying physical processes, depending critically on finite-size, mechanical forces and viscoelastic constraints internal to a pyrenoid, as well as a milieu of crowding, electrostatic and entropic forces originated from the external subcellular environment, such as the spatial patterning of matrix components, all explored theoretically in WP3. WP2.3 will use traditional ensemble level microrheological analysis in vitro to
inform development of bespoke optical tweezer experiments (Fig 4D), further improving their technological capability to enable correlative force manipulation of in situ and reconstituted pyrenoids with simultaneous multispectral SIimfield microscopy. To explore the role of cellular confinement in pyrenoid integrity, we will use dual optical traps to mechanically perturb single reconstituted and native pyrenoids, measuring macroscale changes in morphology at the interface with its local aqueous environment, along with simultaneous multispectral Slimfield to quantify the spatial distribution of fluorescently labelled pyrenoid/matrix components, including protein, membrane and starch components, and the dynamics of their viscoelastic response and determine both protein and
non-protein interaction forces. Correlative super-resolved fluorescence polarization measurements quantify time-resolved changes in orientation of pyrenoid components in response to mechanical perturbation. We will develop protocols to controllably fuse two optically trapped pyrenoids and monitor the time-resolved spatial redistribution of pyrenoid components, and to 'de-fuse' a pyrenoid by pulling it apart into two smaller droplets. We will establish methods to optically trap native pyrenoids in live cells to controllably perturb their spatial location and quantify how their morphology adapts to the boundary constraints of surrounding subcellular structures such as the thylakoid membranes. Each experiment draws on component data from WP1 and develops in communication
with its modelled counterpart in WP3.